The CACNA1A gene is located on chromosome 19p13 and encodes the α1A-sununit of the calcium voltage-gated channel CaV2.1 (P/Q-type). This calcium channels plays a vital role in generating and transmitting electrical signals across the cell by the flow of calcium ions. The calcium channels are located in the presynaptic nerve terminals of central neurons. Calcium flow into the presynaptic terminal causes the release of neurotransmitters, mainly glutamate, into the synaptic cleft.
The different phenotypes caused by a mutation in CACNA1A can partially be explained by the different types of mutation in the gene.
Thus far, all mutations in the CACNA1A gene causing familial/sporadic hemiplegic migraine type 1 (FHM1/SHM1) are amino acid changes in important functional regions. Mutations in CACNA1A causing episodic ataxia type 2 (EA2) are mainly nonsense mutations deletions/insertions disrupting the open reading frame leading to loss-of-function. Mutations in CACNA1A causing spinocerebellar ataxia type 6 (SCA6) are expansions in CAG repeat length. Mutations in CACNA1A causing early infantile epileptic encephalopathy type 42 (EIEE42) are de novo deletions or de novo missense mutations.
Clear genotype-phenotype correlations in patients are almost impossible to make. Therefore, cellular models and animal models offer a solution for better understanding the spectrum and molecular mechanisms.
Cellular models show that an FHM1/SHM1 channels were more likely to open compared to a wild-type channel in response to milder depolarisations and were thus more often in the open position. Thereby increasing the Ca2+ influx and thus increasing the level of neurotransmitter. Knock-in mice show a similar gain-of-function effect when transfected with a human mutation. Taken together it is predicted that mutations in CACNA1A in FHM1/SHM1 lead to increased levels of neurotransmitter in the synaptic cleft of the neurons.
EA2 on the other hand is believed to be caused by a decrease in CaV2.1 channel activity. Although some mouse models show gain-of function instead of loss-of-function.
The functional consequences of a repeat expansion or mutations in CACNA1A leading to respectively SCA6 and EIEE42 have thus far not been elucidated, it is unclear whether it is caused by a dysfunction of CaV2.1 of or in the case of SCA6 due to a toxic gain-of-function effect of the extended glutamine expansion.
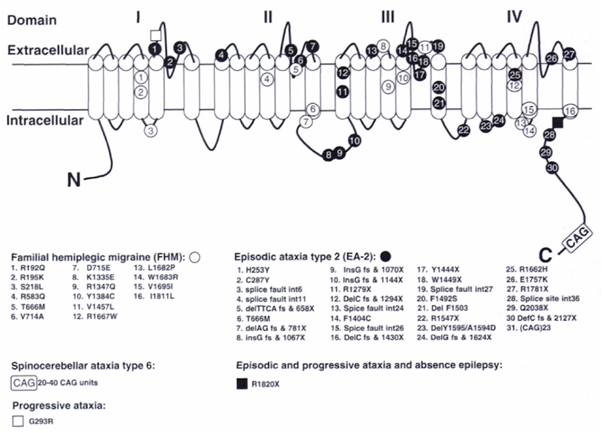
Figure 1: The CACNA1A gene with mutations. The α1A pore-forming subunit of P/Q-type voltage-gated calcium channels is located in the neuron membrane and contains four repeated domains. Each domain includes six membrane-spanning segments and a so-called P-loop between the fifth and sixth segments. Positions of mutations identified in this gene are given, including those identified in mice. 1
1 Ferrari MD, Haan J, Palotie A. (2005). Genetics in Migraine. Oleson J et al. (es.)In The Headaches (3rd edition)(pp. 251-267). Lippincott Williams & Wilkins